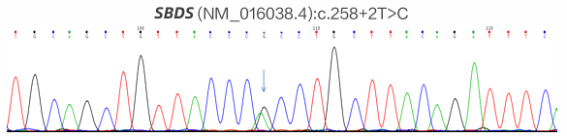
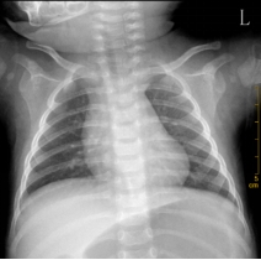
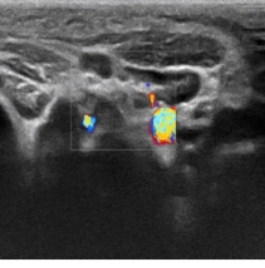
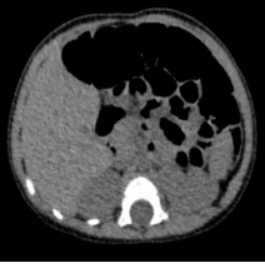
Background: Rare inherited bone marrow failure syndromes pose significant diagnostic challenges in pediatric practice due to their variable and overlapping clinical presentations. Shwachman-Diamond syndrome (SDS) is one such rare autosomal recessive disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, skeletal abnormalities, and variable immune deficiency. Due to its low prevalence and heterogeneous clinical presentation, SDS is frequently underrecognized or misdiagnosed, especially in pediatric patients. Objective: To review the diagnostic and therapeutic process of a pediatric patient with SDS, with the aim of enhancing clinical awareness and understanding of this rare multisystem disease. Methods: A detailed analysis was conducted of the patient’s clinical manifestations, physical examination findings, laboratory results, imaging data, and genetic testing, alongside a review of the therapeutic regimen and follow-up. Results: The patient exhibited hallmark features of SDS, including short stature, recurrent respiratory infections, and persistent neutropenia. Genetic analysis revealed an SBDS (NM_016038.4): c.258+2T>C (intron 2/4) mutation, confirming the diagnosis. Supportive and symptomatic treatments were administered, including infection prevention, nutritional support, and regular monitoring of hematologic status. Conclusion: This case underscores the importance of considering SDS in children with unexplained cytopenias and recurrent infections. Genetic testing plays a pivotal role in achieving a definitive diagnosis. Early recognition and appropriate management can improve outcomes and provide valuable reference for clinicians encountering similar cases.
| Published in | American Journal of Pediatrics (Volume 11, Issue 2) |
| DOI | 10.11648/j.ajp.20251102.15 |
| Page(s) | 58-62 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Shwachman-Diamond Syndrome, Pediatric, Genes
categories | |||||
|---|---|---|---|---|---|
Time | C-reactive protein | White blood cell | Neutrophil | Procalcitonin | Hemoglobin |
Dec. 18 | 29.0 mg/L | 7.90×109/L | 0.07×109/L | 4.15 ng/ml | 94.0 g/L |
Dec. 21 | 11.2 mg/L | 9.56×109/L | 0.83×109/L | 2.45 ng/ml | 95.0 g/L |
Dec. 24 | 8.7 mg/L | 7.10×109/L | 0.15×109/L | 0.29 ng/ml | 92.0 g/L |
Dec. 26 | 2.0 mg/L | 7.62×109/L | 0.48×109/L | 96.0 g/L |
SDS | Shwachman-Diamond Syndrome |
G-CSF | Granulocyte Colony-stimulating Factor |
CT | Computed Tomography |
| [1] | Furutani, E., Liu, S., Galvin, A., et al. (2022). Hematologic complications with age in Shwachman-Diamond syndrome. Blood Advances, 6(1), 297–306. |
| [2] | Hou J. W. (2021). Shwachman‒Diamond syndrome with initial features mimicking common variable immunodeficiency. Pediatrics and neonatology, 62(6), 668–669. |
| [3] | Nelson, A., & Myers, K. (2008). Shwachman-Diamond Syndrome. In M. P. Adam (Ed.), GeneReviews®. University of Washington, Seattle. |
| [4] | Reilly, C. R., & Shimamura, A. (2023). Predisposition to myeloid malignancies in Shwachman-Diamond syndrome: biological insights and clinical advances. Blood, 141(13), 1513–1523. |
| [5] | Farooqui, S. M., Ward, R., & Aziz, M. (2023). Shwachman-Diamond Syndrome. In StatPearls. StatPearls Publishing. |
| [6] | Boocock, G. R., Morrison, J. A., Popovic, M., et al. (2003). Mutations in SBDS are associated with Shwachman- Diamond syndrome. Nature Genetics, 33(1), 97–101. |
| [7] | Han, X., Lu, S., Gu, C., et al. (2023). Clinical features, epidemiology, and treatment of Shwachman-Diamond syndrome: a systematic review. BMC pediatrics, 23(1), 503. |
| [8] | Burwick, N., Coats, S. A., Nakamura, T., et al. (2012). Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood, 120(26), 5143–5152. |
| [9] | Toiviainen-Salo, S., Durie, P. R., Numminen, K., et al. (2009). The natural history of Shwachman-Diamond syndrome- associated liver disease from childhood to adulthood. The Journal of Pediatrics, 155(6), 807–811.e2. |
| [10] | Cesaro, S., Pegoraro, A., Sainati, L., et al. (2020). A prospective study of hematologic complications and long-term survival of Italian patients affected by Shwachman-Diamond syndrome. The Journal of Pediatrics, 219, 196–201. e1. |
| [11] | Thompson, A. S., Giri, N., Gianferante, D. M., et al. (2022). Shwachman Diamond syndrome: narrow genotypic spectrum and variable clinical features. Pediatric research, 92(6), 1671–1680. |
| [12] | Cull, A. H., Kent, D. G., & Warren, A. J. (2024). Emerging genetic technologies informing personalized medicine in Shwachman-Diamond syndrome and other inherited BMF disorders. Blood, 144(9), 931–939. |
| [13] | Peretto, L., Tonetto, E., Maestri, I., et al. (2023). Counteracting the Common Shwachman-Diamond Syndrome- Causing SBDS c.258+2T> C Mutation by RNA Therapeutics and Base/Prime Editing. International journal of molecular sciences, 24(4), 4024. |
| [14] | Cesaro, S., Pillon, M., Sauer, M., et al. (2020). Long-term outcome after allogeneic hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: a retrospective analysis and a review of the literature by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (SAAWP-EBMT). Bone marrow transplantation, 55(9), 1796–1809. |
| [15] | Bezzerri, V., & Cipolli, M. (2019). Shwachman-Diamond syndrome: Molecular mechanisms and current perspectives. Molecular Diagnosis & Therapy, 23(2), 281–290. |
APA Style
Xu, X., Huang, Y. (2025). A Rare Case of Shwachman-Diamond Syndrome: Diagnostic Challenges and Management. American Journal of Pediatrics, 11(2), 58-62. https://doi.org/10.11648/j.ajp.20251102.15
ACS Style
Xu, X.; Huang, Y. A Rare Case of Shwachman-Diamond Syndrome: Diagnostic Challenges and Management. Am. J. Pediatr. 2025, 11(2), 58-62. doi: 10.11648/j.ajp.20251102.15
AMA Style
Xu X, Huang Y. A Rare Case of Shwachman-Diamond Syndrome: Diagnostic Challenges and Management. Am J Pediatr. 2025;11(2):58-62. doi: 10.11648/j.ajp.20251102.15
@article{10.11648/j.ajp.20251102.15,
author = {Xinyi Xu and Yihui Huang},
title = {A Rare Case of Shwachman-Diamond Syndrome: Diagnostic Challenges and Management
},
journal = {American Journal of Pediatrics},
volume = {11},
number = {2},
pages = {58-62},
doi = {10.11648/j.ajp.20251102.15},
url = {https://doi.org/10.11648/j.ajp.20251102.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20251102.15},
abstract = {Background: Rare inherited bone marrow failure syndromes pose significant diagnostic challenges in pediatric practice due to their variable and overlapping clinical presentations. Shwachman-Diamond syndrome (SDS) is one such rare autosomal recessive disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, skeletal abnormalities, and variable immune deficiency. Due to its low prevalence and heterogeneous clinical presentation, SDS is frequently underrecognized or misdiagnosed, especially in pediatric patients. Objective: To review the diagnostic and therapeutic process of a pediatric patient with SDS, with the aim of enhancing clinical awareness and understanding of this rare multisystem disease. Methods: A detailed analysis was conducted of the patient’s clinical manifestations, physical examination findings, laboratory results, imaging data, and genetic testing, alongside a review of the therapeutic regimen and follow-up. Results: The patient exhibited hallmark features of SDS, including short stature, recurrent respiratory infections, and persistent neutropenia. Genetic analysis revealed an SBDS (NM_016038.4): c.258+2T>C (intron 2/4) mutation, confirming the diagnosis. Supportive and symptomatic treatments were administered, including infection prevention, nutritional support, and regular monitoring of hematologic status. Conclusion: This case underscores the importance of considering SDS in children with unexplained cytopenias and recurrent infections. Genetic testing plays a pivotal role in achieving a definitive diagnosis. Early recognition and appropriate management can improve outcomes and provide valuable reference for clinicians encountering similar cases.
},
year = {2025}
}
TY - JOUR T1 - A Rare Case of Shwachman-Diamond Syndrome: Diagnostic Challenges and Management AU - Xinyi Xu AU - Yihui Huang Y1 - 2025/04/29 PY - 2025 N1 - https://doi.org/10.11648/j.ajp.20251102.15 DO - 10.11648/j.ajp.20251102.15 T2 - American Journal of Pediatrics JF - American Journal of Pediatrics JO - American Journal of Pediatrics SP - 58 EP - 62 PB - Science Publishing Group SN - 2472-0909 UR - https://doi.org/10.11648/j.ajp.20251102.15 AB - Background: Rare inherited bone marrow failure syndromes pose significant diagnostic challenges in pediatric practice due to their variable and overlapping clinical presentations. Shwachman-Diamond syndrome (SDS) is one such rare autosomal recessive disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, skeletal abnormalities, and variable immune deficiency. Due to its low prevalence and heterogeneous clinical presentation, SDS is frequently underrecognized or misdiagnosed, especially in pediatric patients. Objective: To review the diagnostic and therapeutic process of a pediatric patient with SDS, with the aim of enhancing clinical awareness and understanding of this rare multisystem disease. Methods: A detailed analysis was conducted of the patient’s clinical manifestations, physical examination findings, laboratory results, imaging data, and genetic testing, alongside a review of the therapeutic regimen and follow-up. Results: The patient exhibited hallmark features of SDS, including short stature, recurrent respiratory infections, and persistent neutropenia. Genetic analysis revealed an SBDS (NM_016038.4): c.258+2T>C (intron 2/4) mutation, confirming the diagnosis. Supportive and symptomatic treatments were administered, including infection prevention, nutritional support, and regular monitoring of hematologic status. Conclusion: This case underscores the importance of considering SDS in children with unexplained cytopenias and recurrent infections. Genetic testing plays a pivotal role in achieving a definitive diagnosis. Early recognition and appropriate management can improve outcomes and provide valuable reference for clinicians encountering similar cases. VL - 11 IS - 2 ER -
Guangzhou Red Cross Hospital of Jinan University, Guangzhou, China
Guangzhou Red Cross Hospital of Jinan University, Guangzhou, China